Tips for Immunohistochemistry
The acceptability of scientific results depends on the accuracy and sensitivity of the appropriate methods used, as well as the controlled specificity of reagents and processes. This is particularly valid for immunohistochemistry (IHC), where sensitivity and specificity of the antibodies and methodological procedures are critical to avoid false-positive and false-negative results.
In IHC, several factors can cause false-positive or false-negative results and all should be verified as much as possible for each experimental setup. The main steps that can lead to false-negative and false-positive results include:
- Detection of the antigen of interest by the primary antibody
- Detection of the primary antibody by secondary antibodies
- Tissue preparation
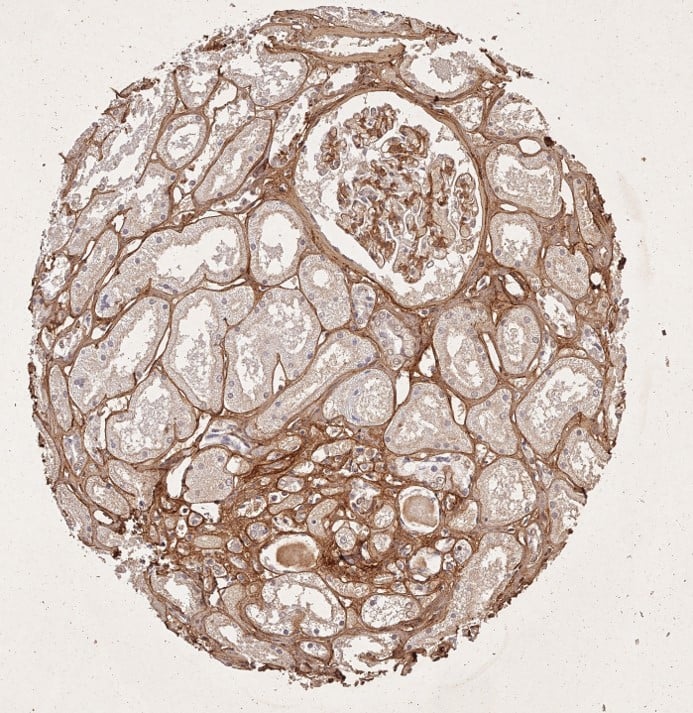
Figure: Immunohistochemistry staining of Collagen Type I.
- Tissue: human tubuli and blood vessels.
- Fixation: FFPE. Antigen
- Retrieval: HIER using Tris-EDTA-citrate buffer pH 7.8 for 5 min.
- Blocking: Peroxidase-Blocking Solution for 10 min.
- Primary Antibody: Anti-Collagen Type I Antibody at 1:15 for 1 hr at 37 °C.
- Secondary Antibody: Dako REAL EnVision Detection Kit, Polymer-HRP, Rabbit/Mouse.
- Counterstain: Hematoxylin for 15 sec.
- Substrate: DAB-Chromogen, Rabbit/Mouse.
- Staining/Results: Intense collagen I staining of fibers surrounding tubuli and around blood vessels.
-
Detecting your Target Protein
Choosing the appropriate IHC controls is an essential step for ensuring the specificity of the primary antibody (i.e. specific antibody-target antigen interaction) and to differentiate between any false positive or false negative results from true signal Primary antibodies can fail to detect their target antigen for many reasons, such as conformation changes induced by fixation/embedding, steric hindrance by interacting proteins/post-translational modifications, low affinity of the antibody for the target, or failure of the antibody to penetrate the tissue. Likewise, antibodies can bind non-specifically to other targets or tissue components.
- Use a positive control—or two antibodies raised against different epitopes of the antigen of interest—as an independent antibody validation control to check for antibody specificity. For a more stringent specificity test, use a negative control, which can be performed by using tissue that is devoid of the antigen of interest (e.g. knockout mouse). Another good control is the blocking/neutralization of the antibody by incubating it with its antigen prior to use for IHC.
-
Detecting your Primary Antibody
Secondary antibodies are raised against immunoglobulins (typically IgGs) of the species in which primary antibodies were raised. Because these antibodies are used in fairly high concentrations, they bind non-specifically to tissue components (extracellular matrix proteins, blood vessels, etc.) instead of the primary antibodies. They might also cross-react with IgGs from other species, which is particularly pertinent to multiple-labeling experiments.
- To minimize cross-reactivity, it is best to use highly pre-adsorbed secondary antibodies.
To test for specificity, secondary antibodies should be applied in the absence of primary antibodies and all residual staining should be considered as non-specific. For example, in immunofluorescence experiments, autofluorescent molecules may be contained in tissues and their presence can be best detected in the absence of secondary antibodies.
- Directly conjugated antibodies should be used only for the detection of very abundant target proteins (e.g. beta-actin and alpha-tubulin).
- For medium to low abundant proteins, use secondary antibodies for detection, as binding of multiple secondary antibodies to a single primary antibody will amplify the signal.
- For very low abundant proteins, use biotinylated secondary antibodies in combination with conjugated avidin/streptavidin, while ensuring endogenous biotin is blocked prior to primary antibody incubation.
- When selecting a fluorophore-conjugated secondary antibody, ensure that the microscope is able to excite and detect the fluorophore appropriately and select photo-stable fluorophores such as DyLight™ Fluorophore dyes and Alexa Fluor® rather than FITC and PE, which are highly susceptible to fading/photo-bleaching. If conducting experiments with multiple fluorescent labels, ensure that each fluorophore can be spectrally separated and that one fluorophore does not get detected in another fluorophore’s channel (a process known as bleed-through).
-
Preparing your Tissue
Relative to antibody specificity, the influence of tissue preparation is more versatile and complex and can result in both false-positive and false-negative results, even when using highly specific and well-characterized antibodies. These effects arise mainly from epitope masking due to fixation-induced conformational changes and failure of the antibody to penetrate the tissue.
Tissue samples can be frozen or fixed. Freezing the sections generally maintains the conformation of the target antigen—allowing superior antibody binding—but small ice crystals that form in the tissue render these sections unsuitable for long-term storage. Fixed and embedded tissue is a better alternative for long-term storage.
Antigens can be masked as a result of the fixation process. The unmasking can be reversed with epitope retrieval/antigen unmasking, which is either mediated by heat (heat-induced epitope retrieval) or proteases (proteolytic-induced epitope retrieval). The latter method acts by degrading the peptides masking the epitope; however, this might also result in alterations to the tissue morphology or the antigen itself. Heat-induced epitope retrieval acts by restoring the secondary and tertiary structure of an epitope and is more frequently used than proteolytic-induced antigen retrieval.
- Follow the antibody supplier recommended antigen retrieval protocol. If no specific protocol is available, use heat-induced antigen retrieval rather than a proteolytic-induced antigen retrieval protocol. For antigen retrieval protocol, use a neutral staining solution. If that does not yield good staining, alkaline or acidic antigen retrieval buffers should be tested.
In addition to pH, other parameters to be optimized are temperature and duration. Blocking should be performed prior to incubation with the primary antibody to prevent non-specific antibody binding. Note that in perfusion-fixed tissue, good results can be obtained without any pre-blocking step.