Lyme Disease (LD) is caused by spirochetal bacteria from the genus Borrelia. Borrelia burgdorferi is the predominant cause of Lyme Disease in the United States, whereas Borrelia afzelii and Borrelia garinii are predominantly implicated in most European incidences. The CDC reported 28,921 confirmed US cases of LD in 2008, presenting a 5% increase of confirmed cases in comparison to 2007. It is estimated that 2.5 million serological LD tests are performed annually.
Recommendations by the CDC for the diagnosis of LD include a two-tiered approach: an initial ELISA-based screening test in conjunction with western blotting to establish the presence of anti-Borrelia antibodies. Currently available LD tests are not only time-consuming but also display a high probability of false-negative and/or positive results. To address this problem, we cloned, expressed, and purified 14 Borrelia-specific antigens for use in the development of a highly sensitive handheld point-of-care (POC) multiplex diagnostic device capable of detecting anti-Borrelia-specific IgM and IgG antibodies in human serum. These 14 antigens, expressed during different stages of the disease, are used to probe LD positive and negative human sera and to raise antigen-specific antibodies. Here we present the preliminary data in our efforts to develop a POC testing device for LD.
Introduction
The current methodology applied in the diagnosis of LD primarily relies on serological applications for the detection of anti-Borrelia antibodies. It is estimated that over 2.5 million LD serology tests are performed annually in the United States; in which a two-tiered approach by ELISA and Western blotting confirms the presence of anti-Borrelia antibodies using a protein cell lysate of an in vitro cultured B. burgdorferi strain.
This process is costly and time-consuming in its entirety and is subjective in the visual evaluation and interpretation of individual test strips. Furthermore, an additional drawback of the current method is that protein cell lysates used for Western blotting contain numerous highly conserved housekeeping proteins that can result in false positive results when detected by antibodies against other bacterial infections. In addition, as B. burgdorferi undergoes rapid adaptive gene expression in response to environmental signals encountered during the different stages of its life cycle in either the arthropod vector or the mammalian host, some proteins are only expressed in the host and not in cell cultures.
To improve upon and simplify the current LD diagnostic approach, we are developing a serological POC assay that may yield results within 10 to 15 minutes. The following are our specific aims:
- Clone, express and purify 14 B. burgdorferi antigens known to be expressed at different stages throughout the infection.
- Evaluate the serological reactivity of human sera from LD patients to define positive and negative assay criteria.
- Lateral flow test development and independent evaluation thereof using defined clinical samples.
Cloning of B. burgdorferi Genes Into E. coli Expression Vectors
The following fourteen (14) proteins were selected as individual components in the flow test device panel: OspA, OspB, OspC, OspE, VlsE, Flagellin, CRASP-1, CRASP-2, DbpA, DbpB, Arp37, surface protein p27, p35 (Bba64) and p39 (BmpA). Respective DNA coding sequences for each protein excluding their export signal and lipidation sequences (hydrophobic N-terminal region) were PCR amplified from B. burgdorferi strain B31 genomic DNA (ATCC Cat# 35210D-5). PCR products were cloned into a modified pMALc2x expression vector (New England Biolabs) to generate respective maltose binding protein (MBP)-antigen fusion proteins using a ligation-independent cloning (LIC) strategy (Figure 1).
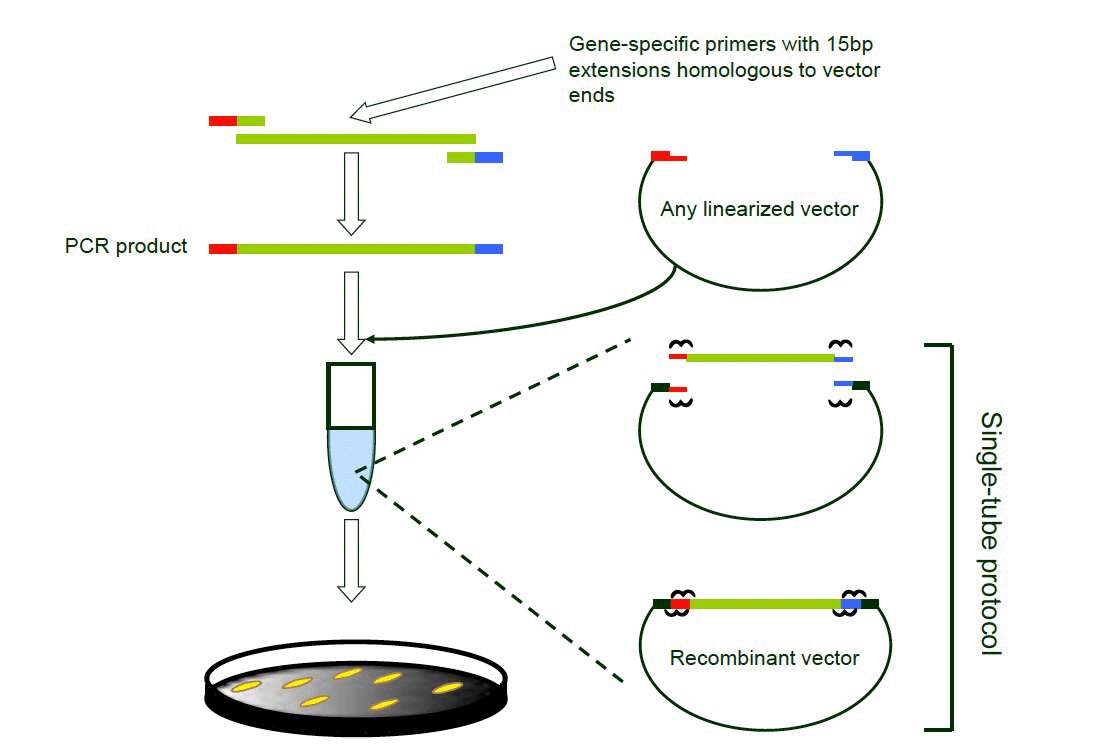
Figure 1. Schematic depicting the ligation-independent cloning (LIC) strategy. A PCR fragment containing 15 base pair overhangs homologous to the vector is cloned into the corresponding linearized vector. Upon homologous recombination, the vector construct is transformed into the E. coli host.
Following transformation, positive clones were identified by restriction analysis before sequencing of the complete DNA insert. Obtained DNA sequence information was analyzed by alignment to respective Genebank B. burgdorferi B31 strain DNA sequences, yielding a 100% homology for all genes with the exception of the DNA sequence encoding VlsE. Three independent VlsE positive clones were sequenced – all of which were identical, but substantially different (94% homology) from the B. burgdorferi B31 VlsE gene (GenBank Accession # BBU76405). The three clones; however, displayed a 100% homology with the VlsE DNA sequence from the European B. burgdorferi sensu lato PBoe strain (GenBank Accession # CAH61549).
Sequence Analysis & Protein Expression

Figure 2. Alignment of three VlsE amino acid sequences. The B. burgdorferi VlsE sequence denoted "Rockland" although the gene was amplified from genomic DNA of strain B31, displays only a 94% homology to the VlsE sequence from strain B31 and a 100% homology to the European strain PBoe.
All 14 plasmids were transformed into the E. coli BL21DE3 pLysS expression host and recombinant MBPfusion protein expression was induced by addition of IPTG (Figure 3A). Expressed fusion proteins were extracted and purified using amylose resin (New England Biolabs, Figure 3B), before individual MBPfusion proteins were cleaved in the presence of the Tobacco Etch Virus (TEV) protease (Figure 3C), thereby releasing the B. burgdorferi recombinant proteins from the MBP. TEV protease was removed by metal affinity chromatography prior to separation of the respective recombinant LD / MBP protein mixture by either size exclusion or ion chromatography (Figure 3D).
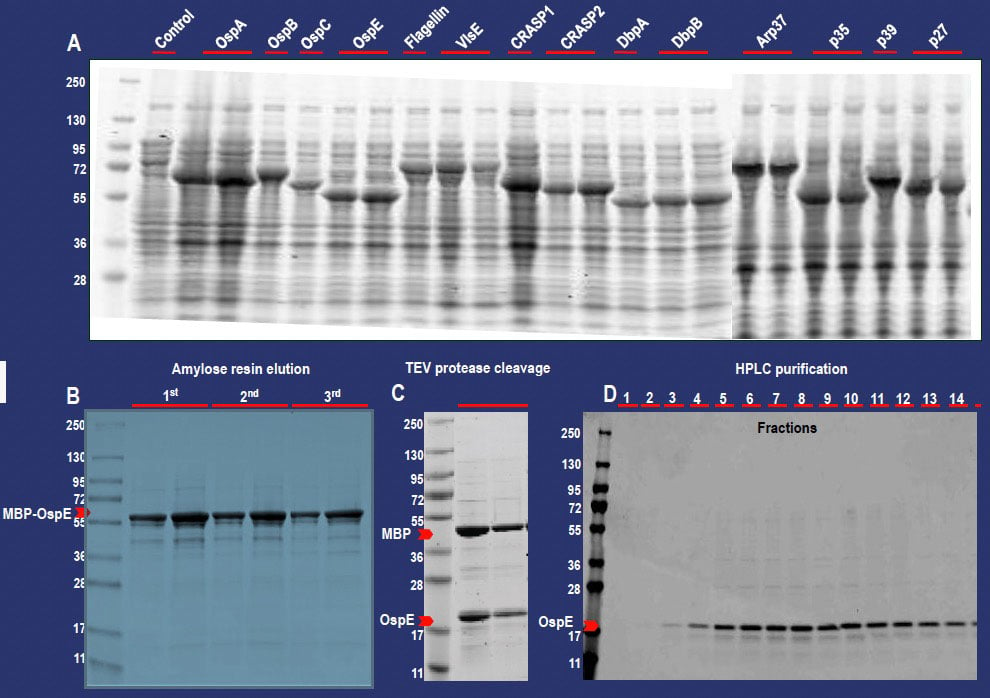
Figure 3. (A) Induced expression of all 14 recombinant LD fusion proteins. Over-expression of the fusion proteins is indicated by the presence of a band not reflected in the control. By way of example, the purification results for MBP-OspE are shown in panels B, C, and D. (B) Elution of the MBP-OspE protein from repeated binding steps to amylose resin. (C) TEV protease-mediated cleavage of the fusion protein, thus releasing the recombinant protein from the MBP. (D) Fraction analysis following MBP and OspE protein separation by SEC.
Lateral Flow Technology
Lateral Flow Technology is well established and forms the basis of numerous strip-based tests including consumer pregnancy tests. Development of a POC strip test for the detection of LD specific antibodies would follow the layout presented above. Anti-LD antibodies present in human whole blood or serum are applied to the sample pad and migrate by capillary action over the gold colloid conjugate pad binding to gold anti-human Ig antibodies present in the pad forming a complex. The complex flows over and binds to respective B. burgdorferi proteins imprinted on the membrane yielding a band indicating the presence of specific circulating antibodies in the specimen. Lateral movement of the antibody complex by capillary action is further promoted by the wick assembly found at the bottom end of the POC testing device.
Proof of Concept Studies
To determine the functionality of a strip-based diagnostic test for the detection of circulating LD specific antibodies, 14 recombinant LD proteins were imprinted onto a nitrocellulose membrane using a slot blotter (Immunetics). In addition, secondary antibody-specific controls (IgG and IgM) as well as MBP were absorbed onto the membrane prior to blocking. Confirmed positive and negative LD human sera were used to bind recombinant proteins followed by the addition of either peroxidase (HRP) conjugated antihuman IgG Fc or anti-human IgM Fc5m secondary antibodies (Figure 4).
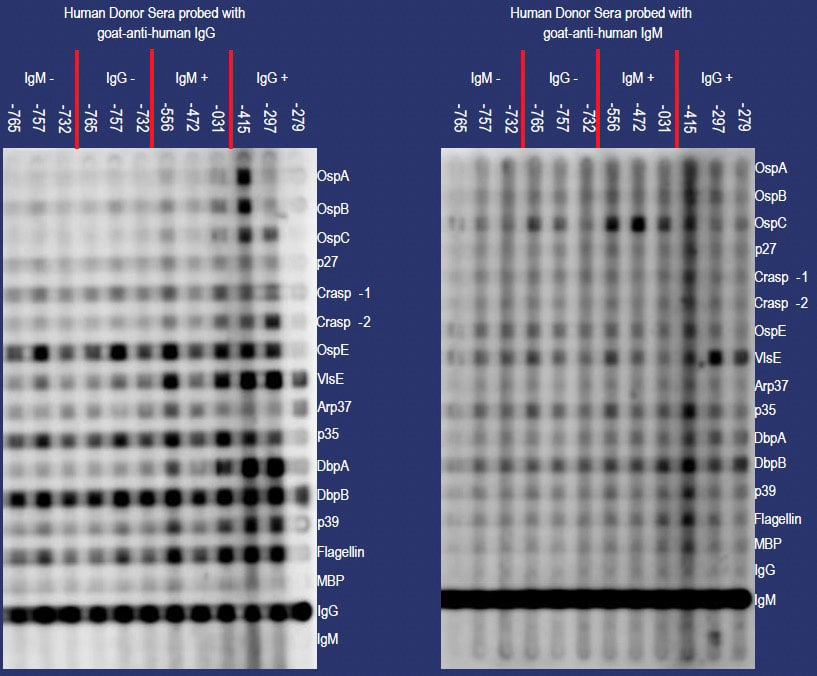
Figure 4. Slot blot matrix resulting from the adsorption and detection of recombinant LD proteins using positive and negative LD human sera. Recombinant proteins were passively absorbed onto the membrane prior to the blocking step. Rotation of the blocked membrane by 90° facilitated the probing of individual proteins with positive and negative LD human sera (indicated by IgG +/- and IgM +/- respectively). IgG and IgM positive controls placed at the bottom of the blot indicate a correctly performed test.
Results indicated a predominantly IgG-based response against various recombinant LD proteins. The strong detection of the recombinant proteins by LD positive human sera is reflected by the intensity of individual blocks in Figure 4. With the exception of OspE, p35, and DbpB which display a strongly reactive pattern in confirmed negative sera, all remaining antigens were only detected by the positive serum. The respective IgG and IgM positive controls included at the bottom of the membrane confirm the validity of the test and also indicate no secondary antibody cross-reactivity. Also, purified MBP is not reactive with human sera as confirmed by the lack of MBP detection shown in Figure 4.
Conclusions & Future Developments
We have successfully expressed all 14 MBP-Borrelia fusion proteins in E. coli. Fusion proteins were effectively cleaved from the MBP carrier protein using Tobacco Etch Virus protease through recognition of its specifically engineered cleavage site. All cleaved B. burgdorferi antigens were subsequently purified by either size exclusion or ion exchange chromatography.
As a Proof-of-Concept we demonstrate that upon imprinting selected recombinant bacterial antigens onto nitrocellulose membrane, LD positive human sera is capable of binding Borrelia-specific proteins, whereas LD negative human sera fails to bind 11 of the 14 antigens. This degree of specificity is sufficient for the further development of a POC lateral flow strip test as proposed.
Future Developments:
- The evaluation of a greater number of well-characterized LD positive and negative human sera in the preliminary format as shown in Figure 4.
- The selection of ten (10) LD indicative markers, including among others the VlsE and DbpA proteins.
- The possible expression and characterization of additional Borrelia proteins.
- The arrangement of recombinant bacterial antigens on the membrane to reflect the bacterial lifecycle expression profile correlating with early, mid, and late stages of infection and validation of expression profiles using LD positive human sera from donors with a well-defined clinical history.
- The manufacture of prototypes including optimization of buffers, ratios of colloidal gold conjugated secondary antibody to human sera, gold colloid size and strip length and validation of prototypes using LD positive human sera from donors representative of early, mid and late-stage infections.
- The independent evaluation of POC lateral flow strip test device prototypes using human clinical sera
References
- Binnicker, M. J., Jespersen, D. J., Harring, J. A., Rollins, L. O., Bryant, S. C., & Beito, E. M. (2008). Evaluation of two commercial systems for automated processing, reading, and interpretation of Lyme borreliosis Western blots. Journal of clinical microbiology
- Goettner, G., Schulte-Spechtel, U., Hillermann, R., Liegl, G., Wilske, B., & Fingerle, V. (2005). Improvement of Lyme borreliosis serodiagnosis by a newly developed recombinant immunoglobulin G (IgG) and IgM line immunoblot assay and addition of VlsE and DbpA homologues. Journal of clinical microbiology
- Hauser, U., Lehnert, G., Lobentanzer, R., & Wilske, B. (1997). Interpretation criteria for standardized Western blots for three European species of Borrelia burgdorferi sensu lato. Journal of clinical microbiology
- Dressler, F., Whalen, J. A., Reinhardt, B. N., & Steere, A. C. (1993). Western blotting in the serodiagnosis of Lyme disease. The Journal of infectious diseases